Alfa- és béta-thalassaemia
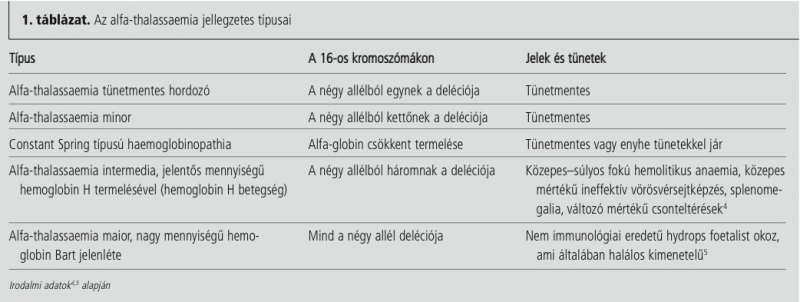
A thalassaemiák (a név a „tenger” görög megfelelőjéből ered) olyan öröklött autoszomális recesszív hematológiai betegségek, melyekben az egyik globin lánc csökkent szintézise vagy hiánya miatt hemolitikus anaemia alakul ki. A globin láncok közti aránytalanság hemolízishez vezet, és gátolja a vörösvérsejtképzést. A családorvosoknak tudniuk kell, hogyan diagnosztizálhatók a thalassaemiák, hogyan különíthetők el az egyéb eredetű microcytaer anaemiától, és hogy milyen lehetőségek vannak a thalassaemia súlyos formáinak kezelésére. Világszerte körülbelül 5%-os gyakorisággal mutathatók ki a globin lánc különféle variánsai, de csak 1,7%-ban mutatható ki alfa- vagy béta-thalassaemia. A thalassaemia egyformán érinti a férfiakat és a nőket, gyakorisága 10 000 élveszületésre számítva körülbelül 4,4. Az alfa-thalassaemia az afrikai és dél-ázsiai származású, a béta-thalassaemia a mediterrán, afrikai és délázsiai származású népességben a leggyakoribb. Ezekben az etnikai csoportokban 5–30%-ban van jelen thalassaemia minor.
A teljes cikket csak regisztrált felhasználóink olvashatják. Kérjük jelentkezzen be az oldalra vagy regisztráljon!
A kulcsos tartalmak megtekintéséhez orvosi regisztráció (pecsétszám) szükséges, amely ingyenes és csak 2 percet vesz igénybe.
a szerző cikkei
James S. Campbell
a szerző cikkei
Prof. dr. Udvardy Miklós, kommentár
a szerző cikkei