A feltételes forgalomba hozatali engedélyezés tapasztalatai
Az EMA nyilvánosságra hozta a feltételes forgalomba hozatali engedélyezési eljárás 10 éves tapasztalatait.

Az egy évig érvényes feltételes forgalomba hozatali engedély (CMA – conditional marketing authorisation) kiadásának célja, hogy a betegek számára hamarabb elérhetővé váljanak olyan gyógyszerek vagy terápiák, amelyek megoldatlan egészségügyi problémák kezelésére lehetnek alkalmasak. Az eljárás 2006 óta létezik, és az Ügynökség a 2006-2016 júniusa közötti adatok felhasználásával tekintette át alkalmazásának előnyeit és hiányosságait körülbelül 40 oldalon. Lássuk a legfontosabb megállapításokat!
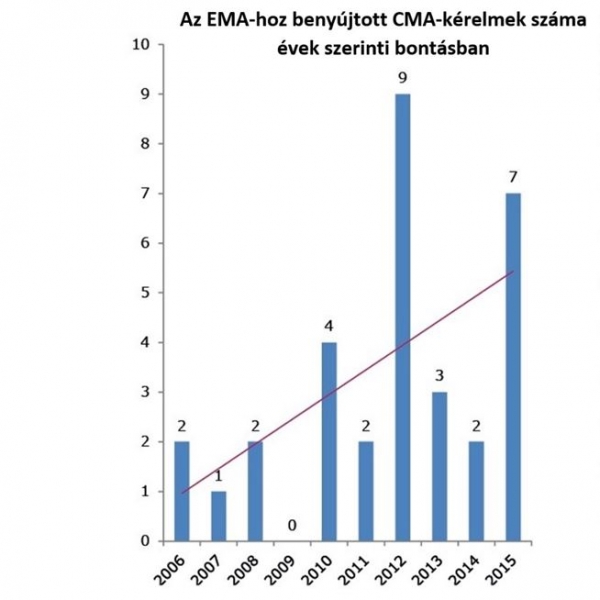
Kialakítása, azaz 2006 óta eltelt 10 évben összesen 30 készítmény kapott ilyen engedélyt.
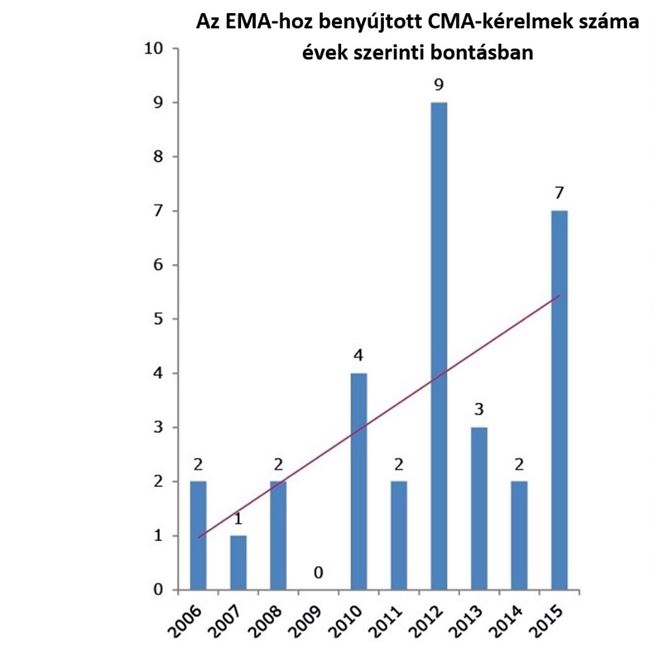
A CMA-t olyan gyógyszerek esetében adja ki az EMA (Európai Gyógyszerügynökség), amelyek súlyos maradandó károsodást, rokkantságot előidéző vagy életveszélyes betegség vagy állapot kezelésében használhatók, mint például a HIV-fertőzés, a mellrák, a súlyos csecsemőkori epilepszia vagy a multirezisztens tuberkulózis. A CMA-val jóváhagyott 30 készítmény közül 14 ritka betegségek kezelésére szolgáló gyógyszer (orphan medicine) volt, mely új terápiás lehetőséget biztosított a ritka betegségekben szenvedő betegeknek. A CMA a betegek korai gyógyszer-hozzáférésének biztosítása érdekében akkor adható ki, ha az adott készítmény azonnali hozzáférésének közegészségügyi előnyei vélhetőleg felülmúlják a gyógyszer hiányos adatokkal történő engedélyezéséből fakadó kockázatokat.
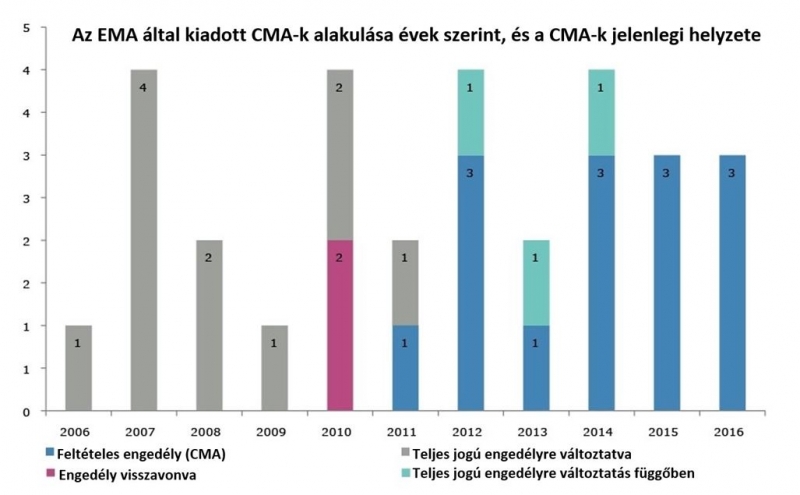
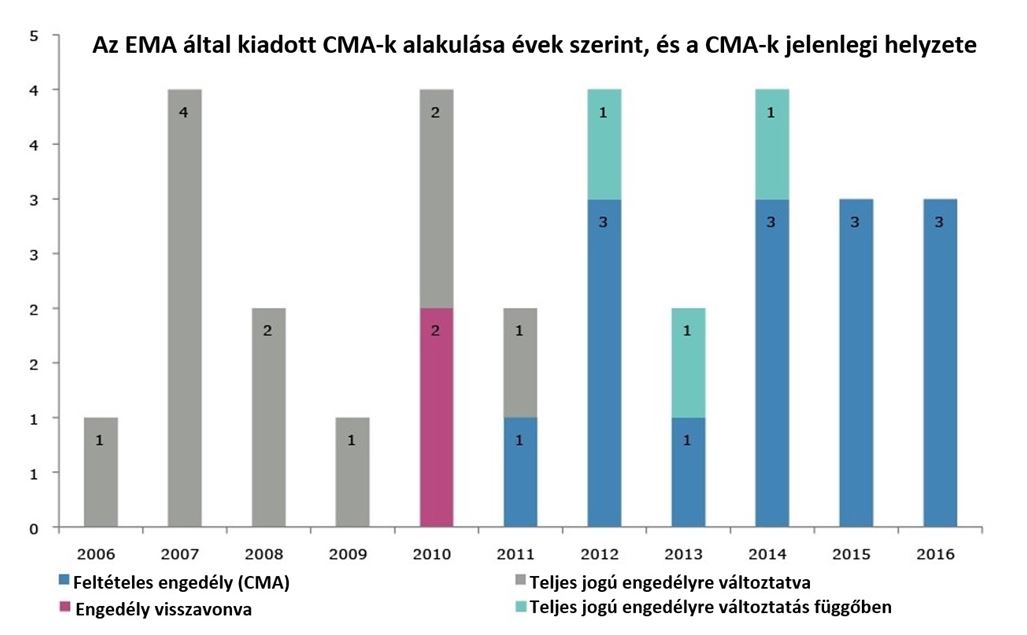
Mivel a CMA csak egy évig érvényes, az eljárás részeként a gyógyszert fejlesztő vállalat kötelezettséget vállal, hogy további vizsgálatokat folytat, és kiegészíti az engedélyezés pillanatában még hiányos adatcsomagot. Az EMA emberi gyógyszerekkel foglalkozó tudományos bizottsága (CHMP) ezeket az engedélyezést követő adatgyűjtést legalább éves gyakorisággal ellenőrzi, és az adatokat áttekinti annak megállapítására, hogy az adott készítmény előny-kockázati profilja továbbra is pozitív maradt-e. Az értékelés végén a CHMP vagy a feltételes forgalomba hozatali engedély meghosszabbítását, vagy visszavonását, vagy standard forgalmazási engedéllyé történő átalakítását javasolja. Az EMA tapasztalatai szerint a forgalomba hozatali engedélyek jogosultjai (MAHs – marketing authorisation holders) igen kevés kivétellel teljesítik az Ügynökség által kiszabott kötelezettségeket. Az esetek több mint 90%-ban a tudományos jellegű kötelezettségek teljesítése nem okoz számottevő változást a profilban, és a különleges kötelezettségek 70%-ánál ezek időtartamát nem is szükséges meghosszabbítani. Átlagosan 4 év telik el a feltételes forgalomba hozatali engedély kiadásától annak „rendes”, azaz teljes jogú forgalomba hozatali engedéllyé konvertálásáig. Ez azt jelenti, hogy a súlyos maradandó károsodást, rokkantságot előidéző vagy életveszélyes betegségekben szenvedő betegek ennyivel korábban érhetik el a számukra kezelési opcióként jelentkező készítményeket.
Bár a feltételes forgalmazási engedélyezési eljárás az elmúlt 10 év tapasztalatai alapján sikeresnek tekinthető, a jelentés megfogalmaz olyan területeket, teendőket, melyek jövőbeni fejlesztésével a program még jobban működhetne:
A CMA-k prospektív jellegű tervezése és az EMA-val folytatott korai konzultáció a magas minőségű adatok generálása érdekében, időben lefolytatott konzultációk a további engedélyezést követő vizsgálatokról és azok kivitelezhetőségéről, valamint a különleges kötelezettségek teljesítése érdekében előírt adatok hatékonyabb beszerzési módjairól.
A betegek számára a gyógyszert hozzáférhetővé tevő egyéb szereplők bevonása a folyamatba, különös tekintettel a HTA (Health Technology Assessment) egészségügyi technológiai értékelő szervezetekre. Ez azért lenne fontos, mert így lenne megállapítható, kijelölhető már a fejlesztési program egy viszonylag korai fázisában a végső döntéshozatalhoz szükséges adatok köre
Az EMA teljes jelentése ezen a linken érhető el.